Research
Surprise in nanoparticle aggregation improves rational design of macromolecular structures
April 01, 2021
Predicting how particles experience changes in charge when they form clusters could markedly improve the accuracy of computational techniques used in drug discovery and in the design of nanomaterials
Accurate prediction of macromolecular structure and function is a key challenge in many fields, from designing new nanomaterials to drug discovery. New counterintuitive findings from Northwestern Engineering could significantly bolster the accuracy of computational structure prediction methods, helping the development of new drugs and nanomaterials.
Erik Luijten and Tine Curk, a postdoctoral researcher in Luijten’s team, found that when two or more identical nanoparticles, proteins, or other biomolecules come together, there’s a spontaneous and global redistribution of charge that can cause initially matching particles to take on different charges in a cluster. The collective effect can qualitatively change the structure of nanoscale systems.

Importantly, this finding shows that charge regulation – the mechanism responsible for the change in particle charge – cannot be ignored when making predictions about how the structure will react.
“This teaches us that global-charge redistribution must be accounted for to accurately predict structure formation of molecular and nanoscale systems,” said Luijten, professor and chair of the materials science and engineering department at the McCormick School of Engineering.
To handle the difficult task of predicting how the changes occur, Curk and Luijten turned to large-scale computer simulations, which showed the surprising results even under very general conditions.
The changing of charges is a complicated, often erratic process. When one particle approaches a second particle the surface charges on both particles change. That then modifies how the new item attracts or repels a third particle, which in turn changes the charge of the first two particles, and so on.
Often, molecules and particles attain charge through dissociation or association of ionic groups on their surface – the same process that governs acid and base reactions. However, this process is perturbed when other charged entities are in the vicinity, such as a DNA molecule that can alter the charge distribution on a nearby protein. Luijten and Curk wondered to what extent these perturbations could add up, leading to possible new modes of interactions and new structures.
They implemented a computer simulation method that dynamically resolves these charge regulation effects, and expected that charge would redistribute within individual particles, as 1950s theoretical arguments proposed.
That was not the case.
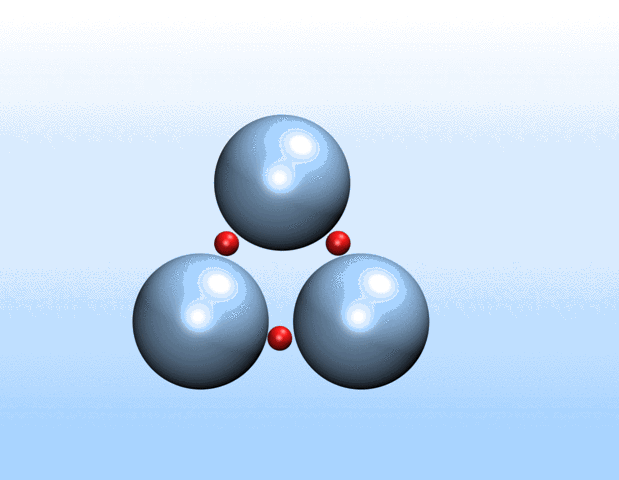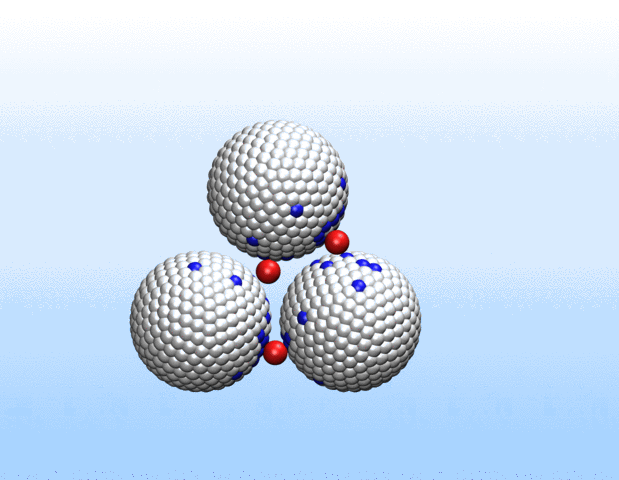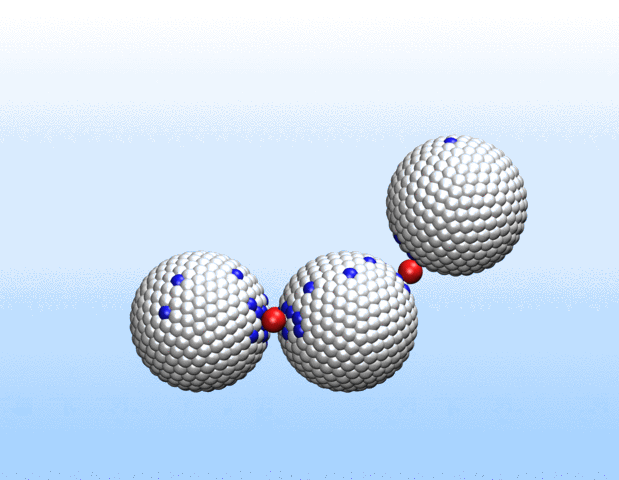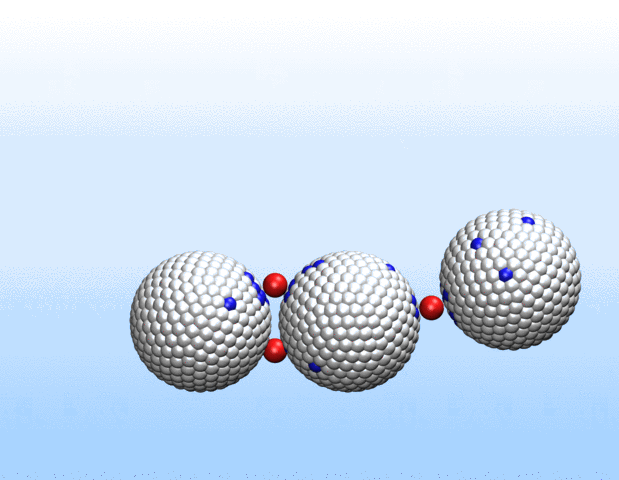
Simulations demonstrated that initially identical nanoparticles spontaneously exchange charge among each other.
“Surprisingly, the simulations demonstrated that initially identical nanoparticles spontaneously exchange charge among each other, resulting in disparate charging and the corresponding formation of asymmetrically shaped clusters,” Curk said.
This charge redistribution process could enable initially identical particles to perform different functions.
“Nanoparticles, proteins, and other biomolecules typically carry electrostatic charges, and this charge controls what types of structures nanoparticles form to create new materials and how proteins function in biological environments,” Luijten said. “Our simulation method has the potential to significantly improve the accuracy of calculations used for drug discovery and the design of new nanomaterials.”
The research paper “Charge Regulation Effects in Nanoparticle Self-Assembly” was published March 31 in the journal Physical Review Letters. Luijten is the Chair of Materials Science and Engineering at Northwestern’s McCormick School of Engineering, as well as Professor of Materials Science and Engineering, Engineering Sciences and Applied Mathematics, and (by courtesy) Physics and Astronomy and Chemistry, and an affiliated faculty member of the International Institute for Nanotechnology.